XLMTM mechanism of disease
Skeletal muscle structure and function are affected by dysfunctional or absent myotubularin protein
Dysfunctional or absent myotubularin protein results in disorganized and defective muscle fibers. This leads to profound muscle weakness and hypotonia, impacting respiratory and neuromuscular motor function.1-4 Myotubularin is a phosphatase involved in several cellular processes required for skeletal muscle maintenance and function, including5-9:
- Muscle growth and differentiation
- Cellular organization and structure
- Cellular function
Normal neuromuscular junction
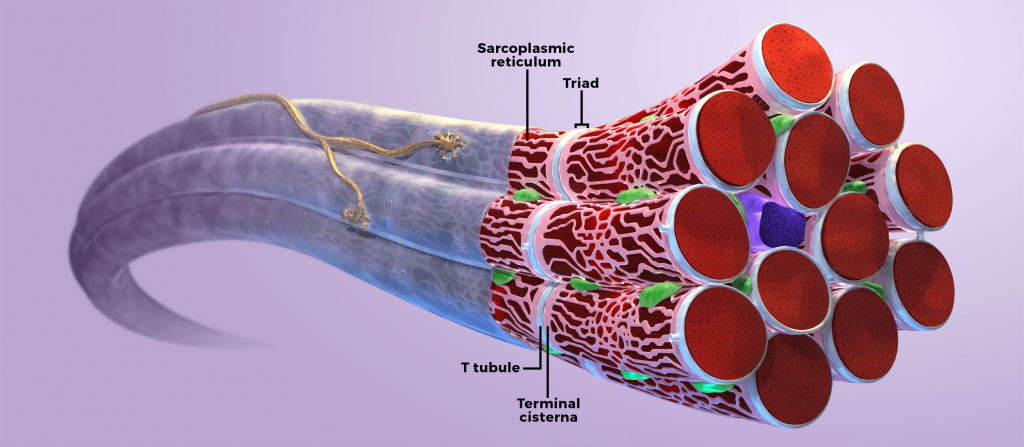
Healthy
In healthy muscle cells, myotubularin protein associates with the muscle triad, which is a structure composed of Tatubules and the terminal sarcoplasmic reticulum.3 The triad mediates excitation-contraction (EC) coupling, a process in which electrical stimuli at the neuromuscular junction (NMJ) are translated into muscle contractions.5
Abnormal
In humans and animal models, defective myotubularin protein in skeletal muscle cells results in impaired EC coupling and mislocalization of cellular organelles. Skeletal muscle impairment, specifically EC coupling, is believed to be a primary cause of profound muscle weakness associated with X-linked myotubular myopathy (XLMTM).4
Understanding XLMTM
Learn about the underlying cause of disease and associated complications.

XLMTM pathology has the potential to be improved
Unlike other neuromuscular disorders, such as Duchenne muscular dystrophy (DMD) or spinal muscular atrophy (SMA), muscle fiber necrosis leading to atrophy is usually absent in XLMTM.10-12 Because the muscle fibers have not atrophied, the skeletal muscle defect in XLMTM is unique in its capacity to be improved by repairing function of the triad and recovering contractile force.3,13-16 Future therapies can potentially improve the pathology of XLMTM.